CoPheScan: Example with Hierarchical Priors
Ichcha Manipur
2026-03-23
Source:vignettes/HierarchicalPriors_04.Rmd
HierarchicalPriors_04.RmdThe hierarchical model is ideal when a large set of variants and phenotypes are available i.e., a phenome-wide association study with a set of variants which are previously known to have a functional role or have been implicated in a disease. Here we show how to use cophescan to infer hierarchical priors on a small test dataset.
data("cophe_multi_trait_data")
trait_dat = cophe_multi_trait_data$summ_stat$Trait_1
str(trait_dat)
#> List of 8
#> $ beta : Named num [1:1000] -0.01369 0.01666 0.09057 -0.00571 -0.05606 ...
#> ..- attr(*, "names")= chr [1:1000] "chr19-11173352" "chr19-11173626" "chr19-11173716" "chr19-11173807" ...
#> $ varbeta: Named num [1:1000] 0.000516 0.000399 0.003124 0.000419 0.000473 ...
#> ..- attr(*, "names")= chr [1:1000] "chr19-11173352" "chr19-11173626" "chr19-11173716" "chr19-11173807" ...
#> $ z : Named num [1:1000] -0.603 0.834 1.62 -0.279 -2.578 ...
#> ..- attr(*, "names")= chr [1:1000] "chr19-11173352" "chr19-11173626" "chr19-11173716" "chr19-11173807" ...
#> $ snp : chr [1:1000] "chr19-11173352" "chr19-11173626" "chr19-11173716" "chr19-11173807" ...
#> $ MAF : Named num [1:1000] 0.2614 0.4871 0.0318 0.4046 0.3042 ...
#> ..- attr(*, "names")= chr [1:1000] "chr19-11173352" "chr19-11173626" "chr19-11173716" "chr19-11173807" ...
#> $ type : chr "cc"
#> $ N : num 20000
#> $ s : num 0.5
querysnpid <- cophe_multi_trait_data$querysnpid
LD <- cophe_multi_trait_data$LDMulti-trait analysis
The first step is preparing the input for the hierarchical model
which are the log Bayes factors: lBF.Ha and lBF.Hc. We will use
cophe.susie.lbf to extract Bayes factors estimated using
SuSIE. Note: When there are no credible sets identified with SuSIE the
function internally calculates lBF.Ha and lBF.Hc using the Approximate
Bayes Factor method.
## Hide print messages from coloc
res.multi.lbf <- list()
for (trait_idx in seq_along(cophe_multi_trait_data$summ_stat)) {
querytrait_ss <- cophe_multi_trait_data$summ_stat[[trait_idx]]
# Here LD is the same
querytrait_ss$LD <- LD
trait_variant_pair <- paste0('Trait', trait_idx, '_', querysnpid)
res.multi.lbf[[trait_variant_pair]] <- cophe.susie.lbf(
querytrait_ss,
querysnpid = querysnpid,
querytrait = paste0('Trait_', trait_idx)
)
}
res.multi.lbf.df = bind_rows(res.multi.lbf)
head(res.multi.lbf.df)
#> lBF.Ha lBF.Hc nsnps querysnp querytrait hit1
#> <num> <num> <int> <char> <char> <char>
#> 1: 15.31003 11.95277 1000 chr19-11182353 Trait_1 chr19-11182353
#> 2: 16.43401 13.00673 1000 chr19-11182353 Trait_2 chr19-11182353
#> 3: 17.36744 13.13794 1000 chr19-11182353 Trait_3 chr19-11182353
#> 4: 14.45423 10.44441 1000 chr19-11182353 Trait_4 chr19-11182353
#> 5: 15.82635 12.54912 1000 chr19-11182353 Trait_5 chr19-11182353
#> 6: 16.23052 12.88257 1000 chr19-11182353 Trait_6 chr19-11182353
#> hit2 typeBF idx1 idx2
#> <char> <char> <int> <int>
#> 1: chr19-11182144 susieBF 1 1
#> 2: chr19-11183133 susieBF 1 1
#> 3: chr19-11189906 susieBF 1 1
#> 4: chr19-11182538 susieBF 1 1
#> 5: chr19-11176397 susieBF 1 1
#> 6: chr19-11182135 susieBF 1 1Note:
- The output of
cophe.susieorcophe.singlecan also be used as input to the hierarchical model as it has all the fields required for the input. This would be useful when you would like to compare results from the fixed priors to those obtained from priors inferred using the hierarchical model. [Swapcophe.susieforcophe.susie.lbfabove and instead of bind_rows do :res.multi.lbf.df = multitrait.simplify(res.multi.lbf)]. - Use the for loop (as above) to run large datasets as storing all the coloc-structured data in a list is memory-intensive. This is also helpful in cases where there are multiple querysnps and different regions of the query traits have to be analysed
Run hierarchical model for priors
The input df for the multi.dat arguments should contain the following fields: “lBF.Ha”,“lBF.Hc” and “nsnps”.
Set the argument covar to TRUE to include covariates
covar = FALSE
covar_vec = rep(1, nrow(res.multi.lbf.df))
## Set covar to TRUE to include covariates, uncomment the following 2 lines
# covar=TRUE
# covar_vec = cophe_multi_trait_data$covar_vec
cophe.hier.res <- run_metrop_priors(
res.multi.lbf.df,
avg_posterior = TRUE,
avg_pik = TRUE,
covar_vec = covar_vec,
covar = covar,
nits = 50000,
thin = 5
)
names(cophe.hier.res)
#> [1] "ll" "parameters" "avg.posterior" "avg.pik"
#> [5] "data" "nits" "thin"Note: Setting posterior or pik to TRUE is memory intensive for very large datasets
Diagnostics of the hierarchical model
Run 3-4 chains of the model and check if there is convergence of chains. Note: For large datasets run the chains separately and save them in individual .RData files. These can be loaded later for diagnostics.
cophe.hier.res.chain.list <- lapply(1:4, function(x) {
run_metrop_priors(
res.multi.lbf.df,
avg_posterior = TRUE,
avg_pik = TRUE,
covar_vec = covar_vec,
covar = covar,
nits = 50000,
thin = 5
)
})
# Store user parameters
old_par <- par(no.readonly = TRUE)
# chain_colors <- c("#e63946c4", "#f1faee", "#a8dadc", "#457b9d" )
chain_colors <- c("#f4f1de", "#e07a5fb2", "#3d405bb2", "#81b29aa6")
layout(
matrix(c(1, 2, 3, 4, 5, 5), ncol = 2, byrow = TRUE),
respect = TRUE,
heights = c(0.9, 0.9, 0.1)
)
matplot(
sapply(cophe.hier.res.chain.list, function(x) x$ll),
type = "l",
col = chain_colors,
main = "loglik",
ylab = "ll",
xlab = "Iteration",
lty = 1
)
y_ax <- c("alpha", "beta", "gamma")
num_pars <- ifelse(covar, 3, 2)
for (idx in 1:num_pars) {
matplot(
sapply(cophe.hier.res.chain.list, function(x) x$parameters[idx, ]),
type = "l",
col = chain_colors,
main = paste(y_ax[idx]),
ylab = y_ax[idx],
xlab = "Iteration",
lty = 1
)
}
if (!covar) {
plot(1, type = "n", axes = FALSE, xlab = "", ylab = "")
}
par(mar = c(0, 0, 0, 0))
plot(1, type = "n", axes = FALSE, xlab = "", ylab = "")
legend(
"top",
legend = paste("Chain", 1:4),
col = chain_colors,
lty = 1,
lwd = 2,
horiz = TRUE,
bty = "n"
)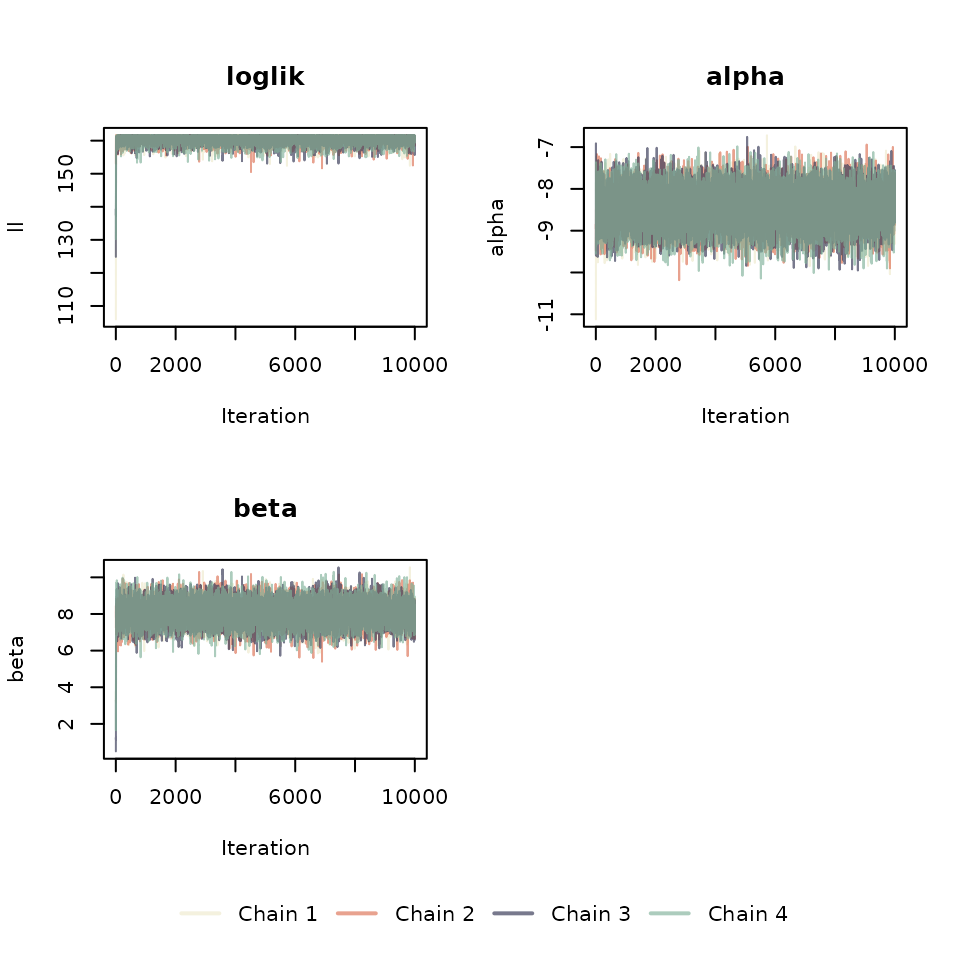
# Reset user parameters
par(old_par)Prediction
cophe.hier.res.chain.list[[1]]$avg.posterior contains
the posterior probabilities of the hypotheses :
,
and
for the queryvariant/querytrait pairs obtained from the hierarchical
model. Here we use the first chain for prediction.
res.post.prob = cbind(
cophe.hier.res.chain.list[[1]]$avg.posterior,
cophe.hier.res$data
)We can use the cophe.hyp.predict function to predict the
hypothesis given the posterior probabilities. The cophe.hyp.call column
shows the predicted hypothesis for each query trait-query variant
pair.
res.hier.predict <- cophe.hyp.predict(as.data.frame(res.post.prob))
#> Hc.cutoff = 0.6
#> Hn.cutoff = 0.2
col_disp <- c(
"PP.Hn",
"PP.Ha",
"PP.Hc",
"nsnps",
"querysnp",
"querytrait",
"typeBF",
"grp",
"cophe.hyp.call"
)
knitr::kable(
res.hier.predict[, col_disp],
row.names = FALSE,
digits = 3,
align = "c"
)| PP.Hn | PP.Ha | PP.Hc | nsnps | querysnp | querytrait | typeBF | grp | cophe.hyp.call |
|---|---|---|---|---|---|---|---|---|
| 0.772 | 0.099 | 0.128 | 1000 | chr19-11182353 | Trait_17 | ABF | chr19-11182353_Trait_17 | Hn |
| 0.790 | 0.073 | 0.137 | 1000 | chr19-11182353 | Trait_18 | ABF | chr19-11182353_Trait_18 | Hn |
| 0.797 | 0.083 | 0.119 | 1000 | chr19-11182353 | Trait_19 | ABF | chr19-11182353_Trait_19 | Hn |
| 0.800 | 0.073 | 0.127 | 1000 | chr19-11182353 | Trait_20 | ABF | chr19-11182353_Trait_20 | Hn |
| 0.693 | 0.058 | 0.250 | 1000 | chr19-11182353 | Trait_21 | ABF | chr19-11182353_Trait_21 | Hn |
| 0.656 | 0.077 | 0.266 | 1000 | chr19-11182353 | Trait_22 | ABF | chr19-11182353_Trait_22 | Hn |
| 0.789 | 0.081 | 0.130 | 1000 | chr19-11182353 | Trait_23 | ABF | chr19-11182353_Trait_23 | Hn |
| 0.806 | 0.074 | 0.120 | 1000 | chr19-11182353 | Trait_24 | ABF | chr19-11182353_Trait_24 | Hn |
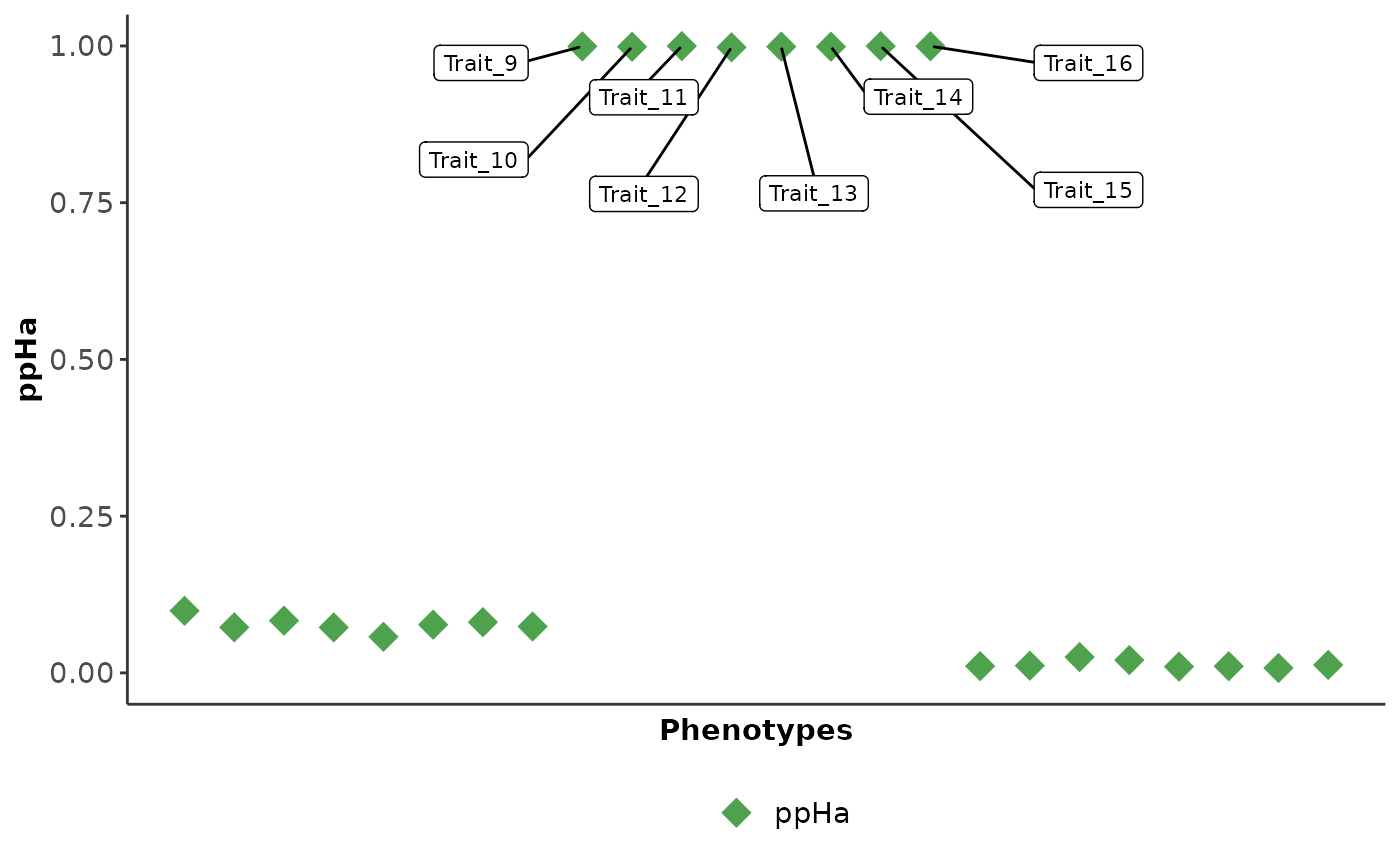
| 0.000 | 0.999 | 0.000 | 1000 | chr19-11182353 | Trait_9 | susieBF | chr19-11182353_Trait_9 | Ha |
| 0.001 | 0.999 | 0.000 | 1000 | chr19-11182353 | Trait_10 | susieBF | chr19-11182353_Trait_10 | Ha |
| 0.000 | 1.000 | 0.000 | 1000 | chr19-11182353 | Trait_11 | susieBF | chr19-11182353_Trait_11 | Ha |
| 0.002 | 0.998 | 0.000 | 1000 | chr19-11182353 | Trait_12 | susieBF | chr19-11182353_Trait_12 | Ha |
| 0.001 | 0.999 | 0.000 | 1000 | chr19-11182353 | Trait_13 | susieBF | chr19-11182353_Trait_13 | Ha |
| 0.000 | 0.999 | 0.000 | 1000 | chr19-11182353 | Trait_14 | susieBF | chr19-11182353_Trait_14 | Ha |
| 0.000 | 1.000 | 0.000 | 1000 | chr19-11182353 | Trait_15 | susieBF | chr19-11182353_Trait_15 | Ha |
| 0.000 | 1.000 | 0.000 | 1000 | chr19-11182353 | Trait_16 | susieBF | chr19-11182353_Trait_16 | Ha |
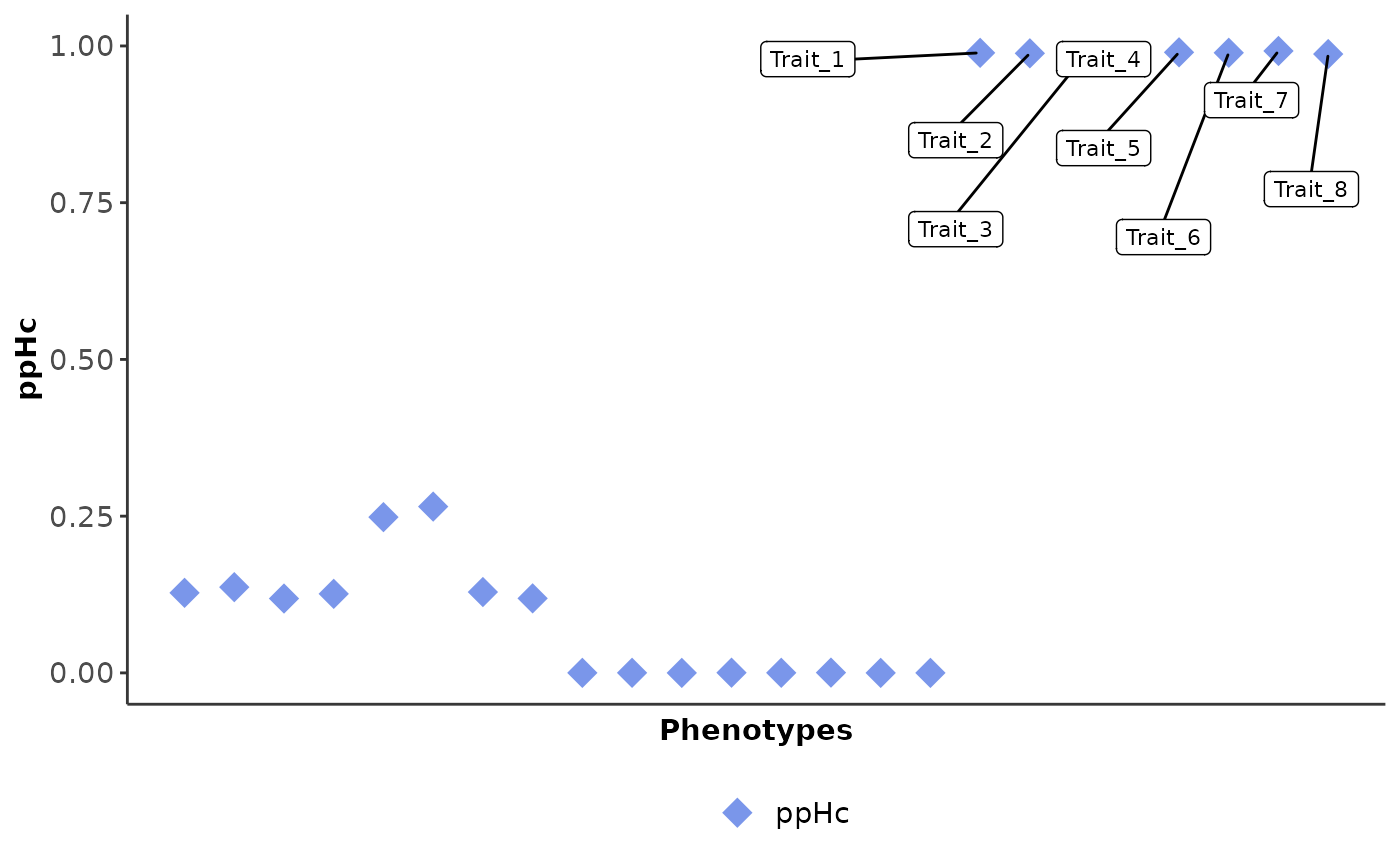
| 0.000 | 0.011 | 0.989 | 1000 | chr19-11182353 | Trait_1 | susieBF | chr19-11182353_Trait_1 | Hc |
| 0.000 | 0.011 | 0.988 | 1000 | chr19-11182353 | Trait_2 | susieBF | chr19-11182353_Trait_2 | Hc |
| 0.000 | 0.025 | 0.975 | 1000 | chr19-11182353 | Trait_3 | susieBF | chr19-11182353_Trait_3 | Hc |
| 0.000 | 0.020 | 0.979 | 1000 | chr19-11182353 | Trait_4 | susieBF | chr19-11182353_Trait_4 | Hc |
| 0.000 | 0.010 | 0.990 | 1000 | chr19-11182353 | Trait_5 | susieBF | chr19-11182353_Trait_5 | Hc |
| 0.000 | 0.011 | 0.989 | 1000 | chr19-11182353 | Trait_6 | susieBF | chr19-11182353_Trait_6 | Hc |
| 0.000 | 0.008 | 0.992 | 1000 | chr19-11182353 | Trait_7 | susieBF | chr19-11182353_Trait_7 | Hc |
| 0.000 | 0.013 | 0.987 | 1000 | chr19-11182353 | Trait_8 | susieBF | chr19-11182353_Trait_8 | Hc |
Visualisation
Use the cophe_plot function to return -log10(pval), ppHa
and ppHc PheWAS plots from the cophescan output.
res.plots = cophe_plot(
res.hier.predict,
traits.dat = cophe_multi_trait_data$summ_stat,
querysnpid = querysnpid,
query_trait_names = paste0('Trait_', 1:24)
)
# if (!require(ggpubr)) {
# install.packages("ggpubr")
# }
# ggpubr::ggarrange(res.plots$pval, res.plots$ppHa, res.plots$ppHc, ncol = 2,
# nrow = 2)
Note: For large datasets, it’s not feasible to input all
coloc-structured data into “traits.dat” at once. Instead, use a loop and
run the “get_beta” function over all the trait-variant pairs, and
provide the resulting data frame (after binding the rows) as the
“beta_p” argument in cophe_plot:
# beta_p_list <- lapply(seq_along(cophe_multi_trait_data$summ_stat), function(x) get_beta(list(cophe_multi_trait_data$summ_stat[[x]]), querysnpid, names(cophe_multi_trait_data$summ_stat)[x]))
# ### the datasets need not be in a list as in cophe_multi_trait_data$summ_stat and can be stored independently.
# beta_p_df = bind_rows(beta_p_list)
# ### Make sure the query trait names in beta_p_df are the same as in res.hier.predict
# res.plots = cophe_plot(res.hier.predict, querysnpid = querysnpid, query_trait_names = beta_p_df$querytrait, beta_p = beta_p_df)